AlphaFold2阅读笔记
AlphaFold2阅读笔记
背景

AF2给计算生物学及生物医药行业带来了可以说是巨大的影响,因为它确实解决或者说极大的推动了蛋白结构领域的发展。他带动的下游生物医药产业链,各个公司及课题组布局不计其数,未来5年内应该有望保持相当的热度。所以很有必要好好的了解其core idea。
总体框架
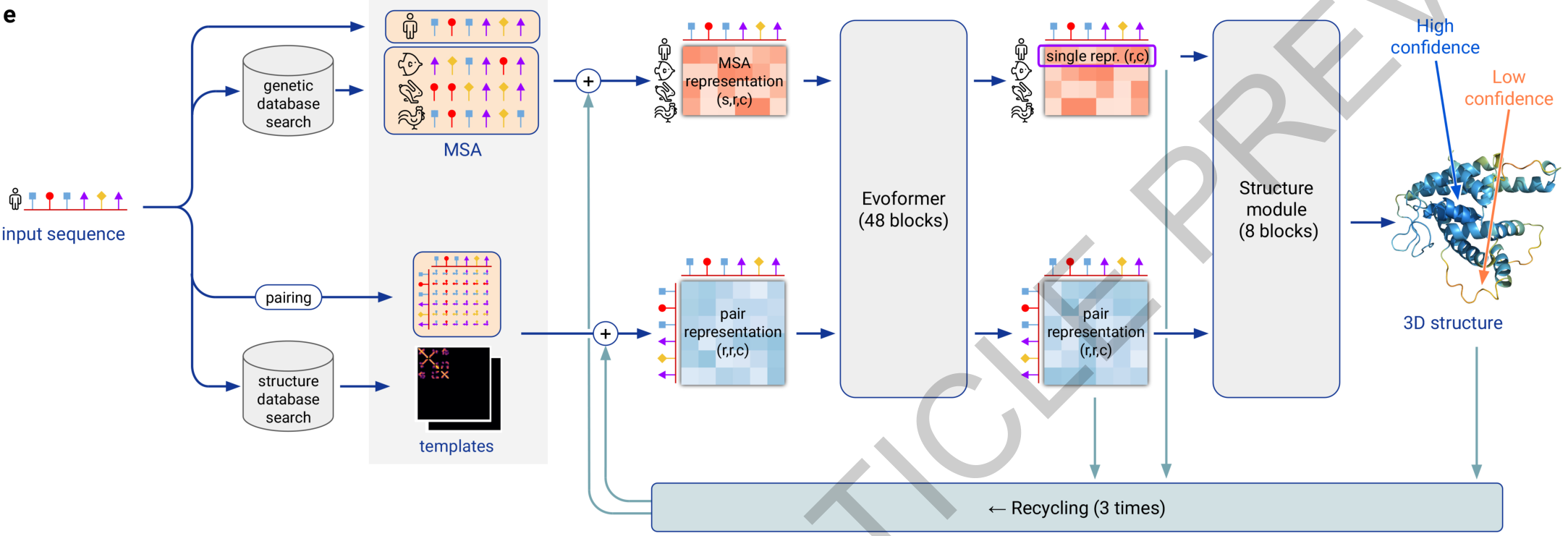
从总体框架上可以看出,其输入主要包含两个方面:多序列比对(MSA)和已有的蛋白结构。使用已有的结构很容易理解,因为类似于同源建模,少量的序列改变大多数情况不会对结构造成根本性的改变,当然,不排除一些低概率下确实会导致结构很大变化,那极有可能是变化的那少数氨基酸极大的改变了蛋白folding的自由能面。另一个使用的是MSA,虽然近年结构出产越来越快,不过比起已知的序列来说,仍然是九牛身上的一根毛,所以能将已知的序列信息利用起来那自然是极好的,使用MSA的核心理论是欧式空间上相近的氨基酸进化上有更大概率是同步突变的,同时,不同物种间同源蛋白常有相似的结构但不同的序列,挖掘这些信息已经被证明对预测结构十分有效。
再来看模型主体,很明显,一个蛋白有两种表示方法,一维的和二维的。一维的表示以序列来源的信息为主,而二维的表示则以结构来源的信息为主。粗暴点说,一种是序列表示,另一种是序列的成对表示。其中,它使用了一种原创的evoformer来同时更新这两种蛋白表示,最后通过一个推断模块可视化结构。除此之外,其还有recycle的成分,这些都在下文介绍。
模型输入
其中包括一维的和二维的表示,包括MSA序列及结构特征。
MSA序列的特征
| msa_feat | $ [N_{clust}, N_{res}, 49] $ | 内容 |
|---|---|---|
| cluster_msa | $ [N_{clust}, N_{res}, 23] $ | 20种残基+unknown+gap+mask的 |
| cluster_has_deletion | $ [N_{clust}, N_{res}, 1] $ | cluster上该位置氨基酸是否有删除(msa来源的数据中,align后会删除预测序列的gap,同时删除aligned其他序列相应位置) |
| cluster_deletion_value | $ [N_{clust}, N_{res}, 1] $ | 该cluster中心删除数目 |
| cluster_deletion_mean | $ [N_{clust}, N_{res}, 1] $ | 该cluster中心删除数目均值 |
| cluster_profile | $ [N_{clust}, N_{res}, 23] $ | 该cluster的23类残基的分布 |
| extra_msa_feat | $ [N_{extra_seq}, N_{res}, 25] $ | 内容 |
|---|---|---|
| extra_msa | $ [N_{extra_seq}, N_{res}, 23] $ | 20种残基+unknown+gap+mask的 |
| extra_msa_has_deletion | $ [N_{extra_seq}, N_{res}, 1] $ | 是否有删除(同上理由,删除目标序列gap,同时其他序列对应位置) |
| extra_msa_deletion_value | $ [N_{extra_seq}, N_{res}, 1] $ | 删除数目 |
这个MSA来源的feature很容易理解,举个例子如下:

将第一行的序列当做目标序列,假设得到了如图的MSA。首先第一步是去除目标序列的gap,同时其余序列的对应位置也剪切,deletion相关的feature即这么来的。
cluster center是任意选出的$ N_{clust} $条序列,其余序列根据汉明距离汇聚到中心上,其中第一条center默认为目标序列,由此得到的数据为msa_feat,剩余聚类center以外的成为extra_msa_feat。
target_feat:$ [N_{res}, 21] $,20种残基+unknown。
residue_index:$ [N_{res}] $,index表示残基位置。
结构的特征
| template_angle_feat | $ [N_{templ}, N_{res}, 51] $ | 内容 |
|---|---|---|
| template_aatype | $ [N_{templ}, N_{res}, 22] $ | 20种残基+unknown+gap |
| template_torsion_angles | $ [N_{templ}, N_{res}, 14] $ | 主链3个内坐标角度,支链最多达4个内坐标角度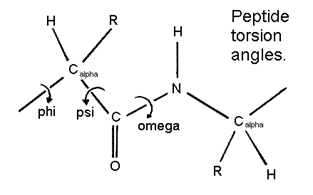 |
| template_alt_torsion_angles | $ [N_{templ}, N_{res}, 14] $ | template_torsion_angles的对称角度表示 |
| template_torsion_angles_mask | $ [N_{templ}, N_{res}, 14] $ | 内坐标角度的mask |
Mask
AF2的对MSA的mask力度很大,类似BERT,即15%产生如下mask:
- 10% 残基变为任意残基
- 10% 残基变为msa同位置上任意残基
- 10% 残基不变
- 70% 残基变为一个 special token(masked_msa_token)
结构template的mask仅用于该残基位置是否有坐标信息(Template search找到的结果可能只匹配部分序列)
Input feature embeddings
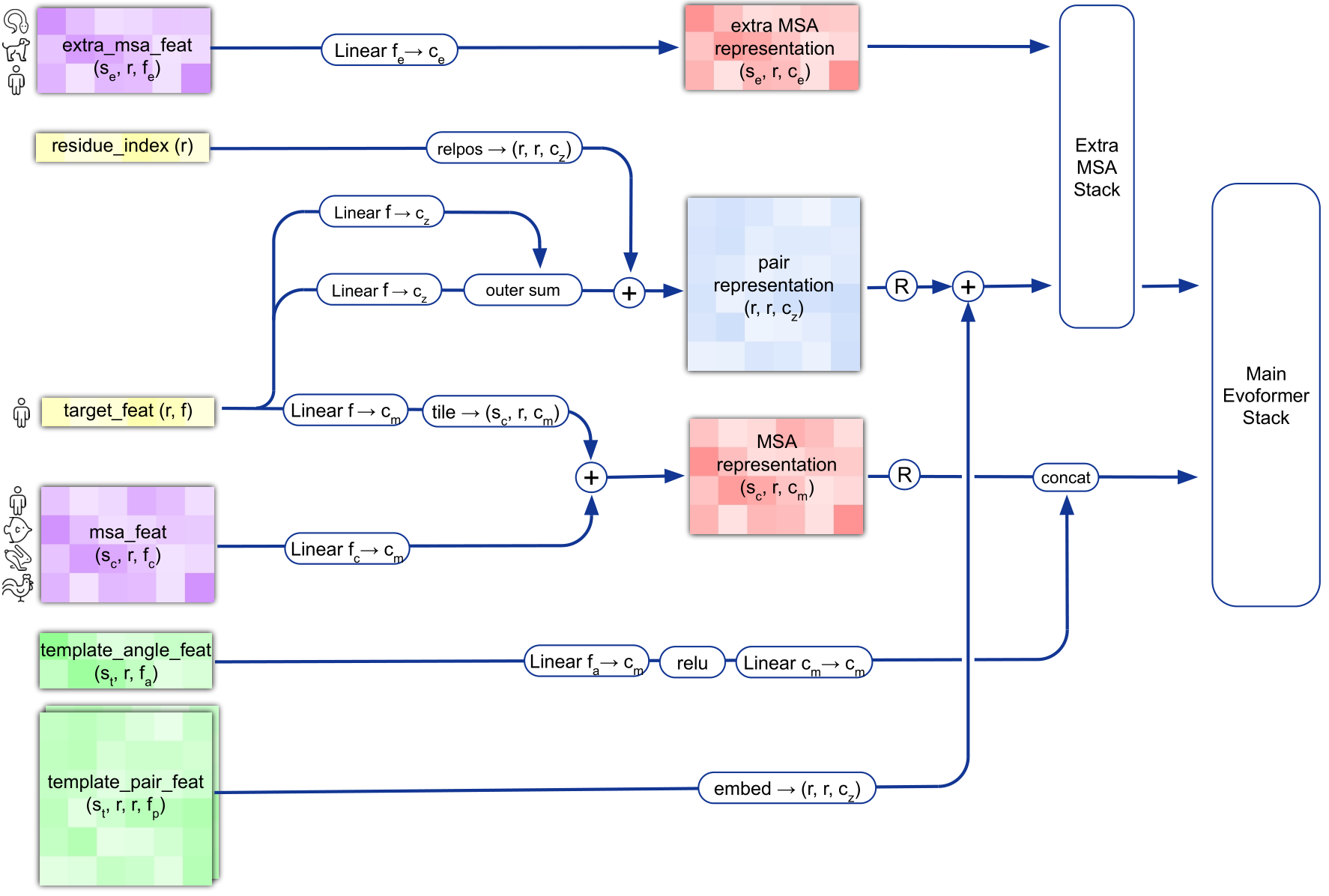
图表示的十分清晰了。extra_msa_feat是一个非常大的张量,仅作为主要msa_feat的补充,使用一个与Main Evoformer类似但channel比较小的模块处理后送入模型主体。
值得注意的是,图中R代表recycle,每个cycle会重新选取cluster中心。同时,每次recycle都采用ensemble的模式($ N_{ensemble} $),模型每个cycle的结果取平均,不过文中说 $ N_{ensemble}=1 $ 时也有较好效果。同时recycle时是把前个cycle的结构输出的$ C{β} $间分箱的距离矩阵加入到后一个cycle的输入。
Evoformer

Evoformer本质是Transformer的变体。其中红色模块代表attention相关的变换,绿色代表简单的Feed Forward,黄色代表“三角更新”(其文中认为是attention的下位替代,用于减少计算开销)。
MSA repr.更新
其中主要是对其第一维和第二维作attention
Row-wise gated self-attention with pair bias
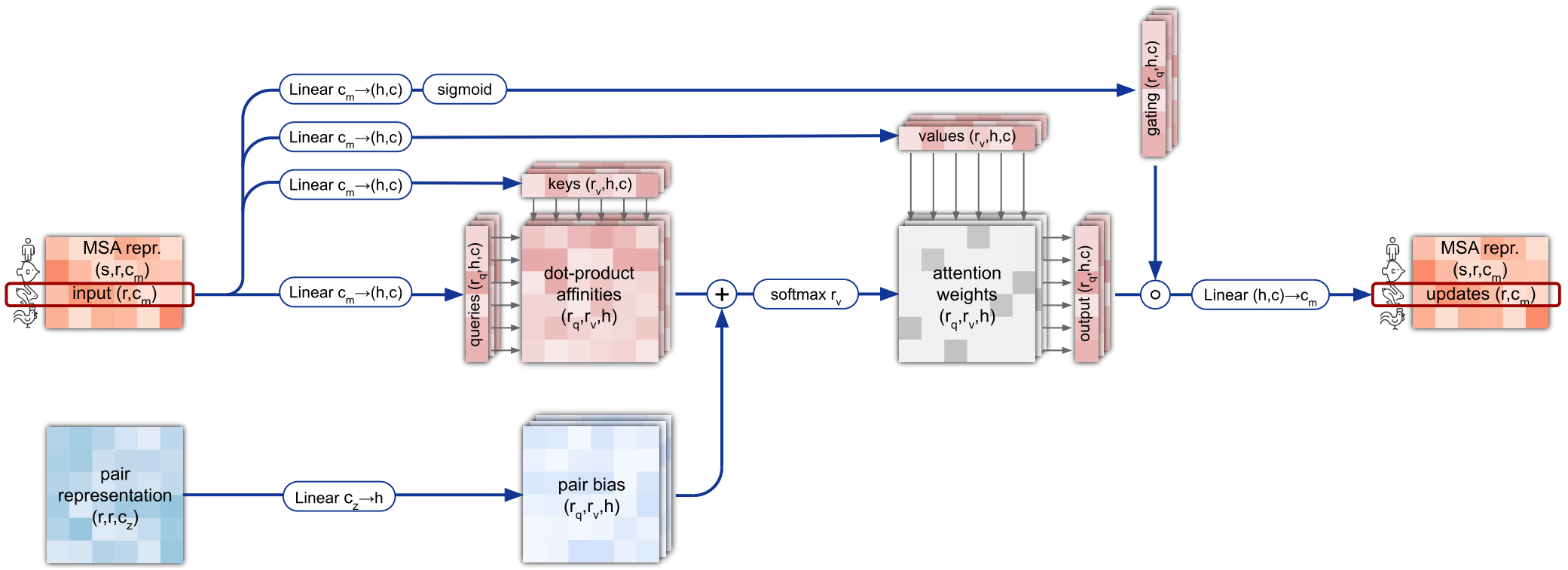
简言之,对第一维即$ N_{clust} $的每条序列进行self-attention,不过使用的是linear不带bias,bias由pair repr.组成。除此外还有个gate,结合直接连接的残差结构决定更新的程度。
Column-wise gated self-attention
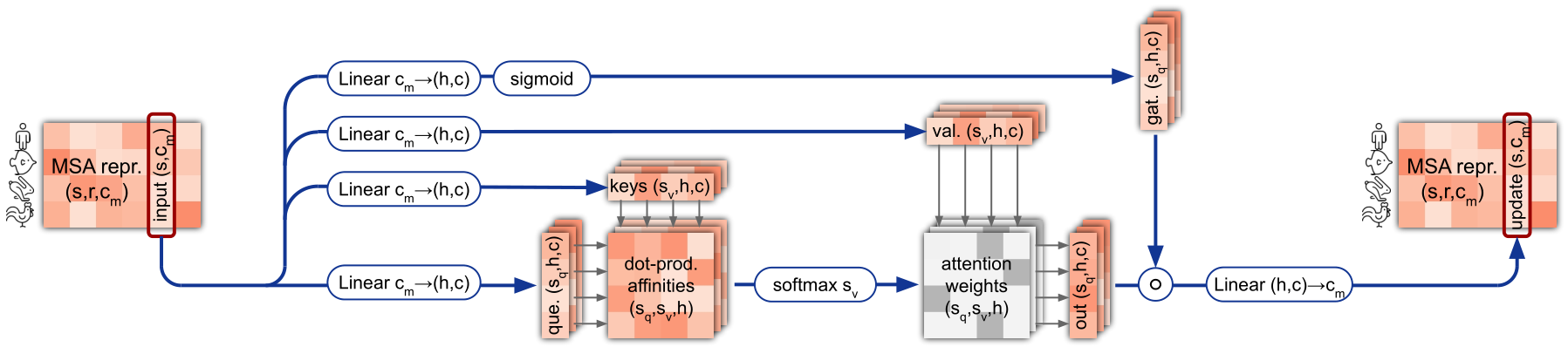
常规attention,对第二维即$ N_{res} $的每条序列进行self-attention。
Transition layer
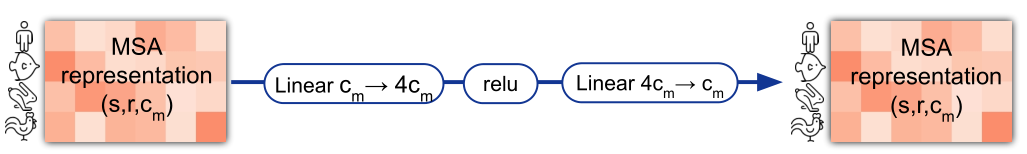
简言之,一个两层MLP。
pair-wise repr.更新
其涉及对第一维及第二维的三角更新及attention
MSA to pair-wise repr.
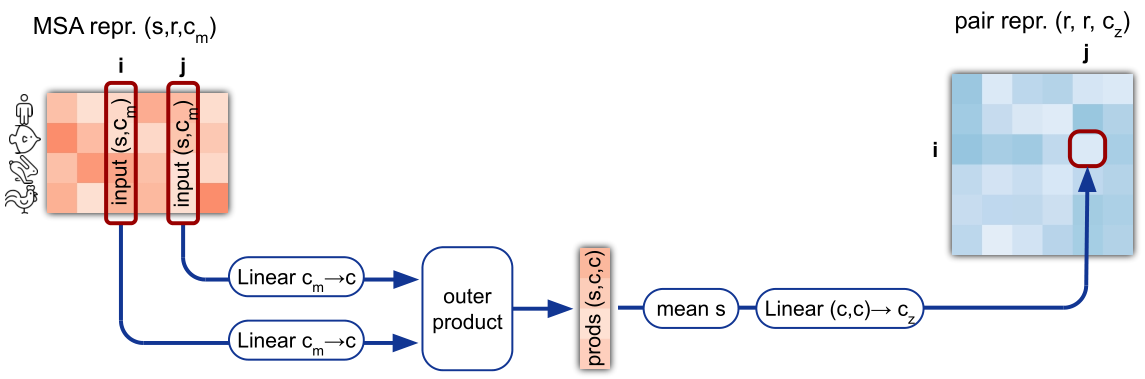
使用外积将一维数据变为二维数据,不过由于一维的表示多了个$ N_{clust} $,所以在这一维度取均值表示。
Triangular multiplicative update
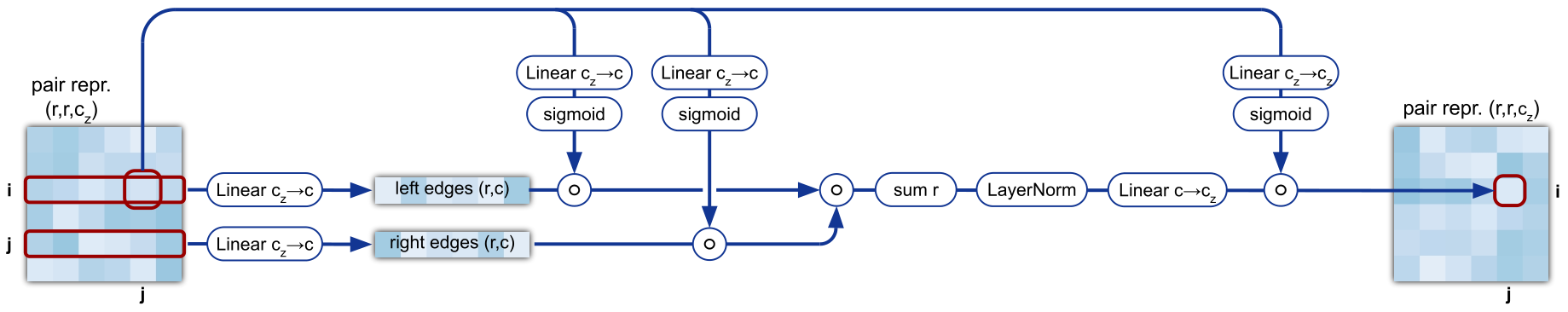
作为attention的下位替代,其文中说单独使用也有不错的效果。两个三角更新模块分别是对第一维(outgoing)和第二维(incoming)使用。从示意图及伪代码上可以看出,其比正常的attention的Q,K,V要少一个,粗暴理解的话,可以认为是仅有Q与K相乘,然后直接sum再norm。
Triangular self-attention
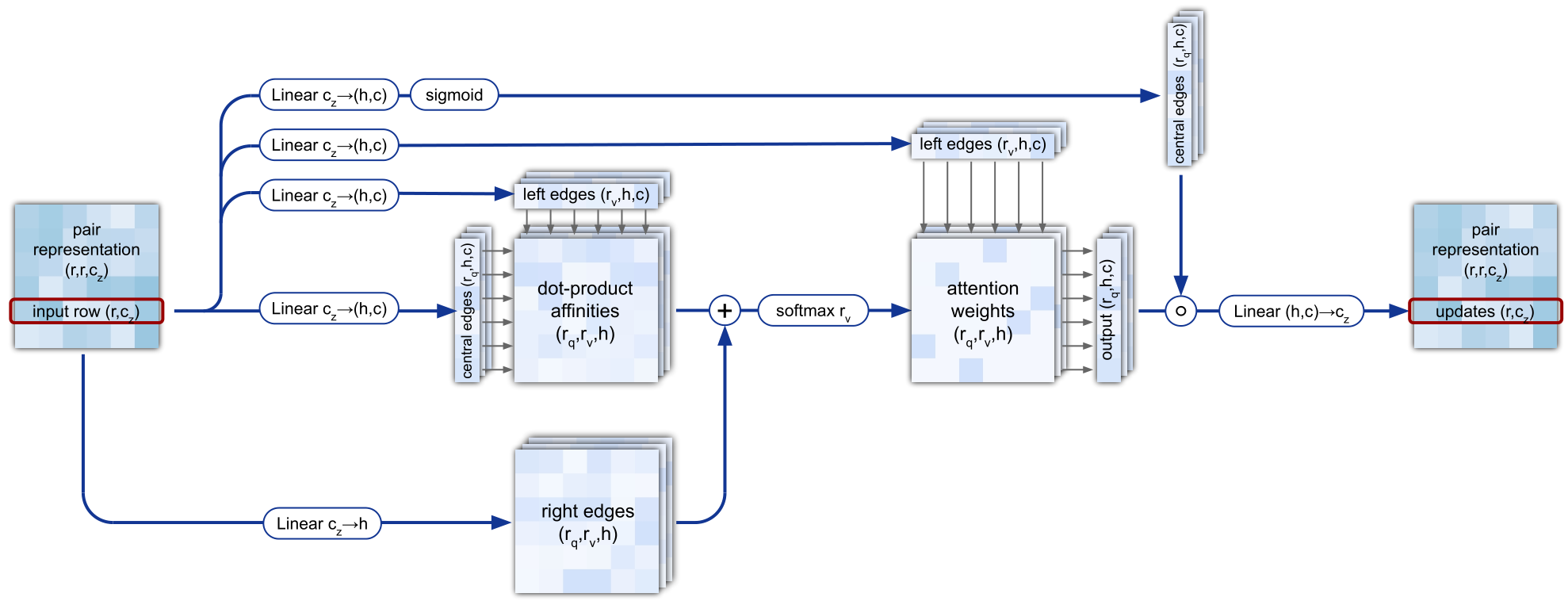
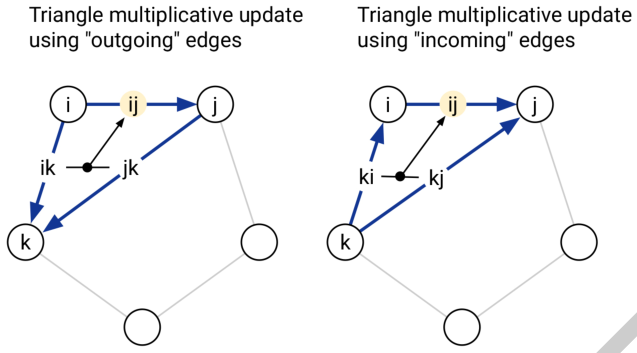
Triangular multiplicative update与Triangular self-attention模块的本质是将蛋白序列看做一张全连接的GNN,而Triangular self-attention与GAT非常相似。如果资源足够,可能甚至不用根据维度作attention,而是在全局作attention,不过那样张量的会变得不切实际的大。
Transition
简言之,一个两层MLP。
推断模块(Invariant Point Attention Module)
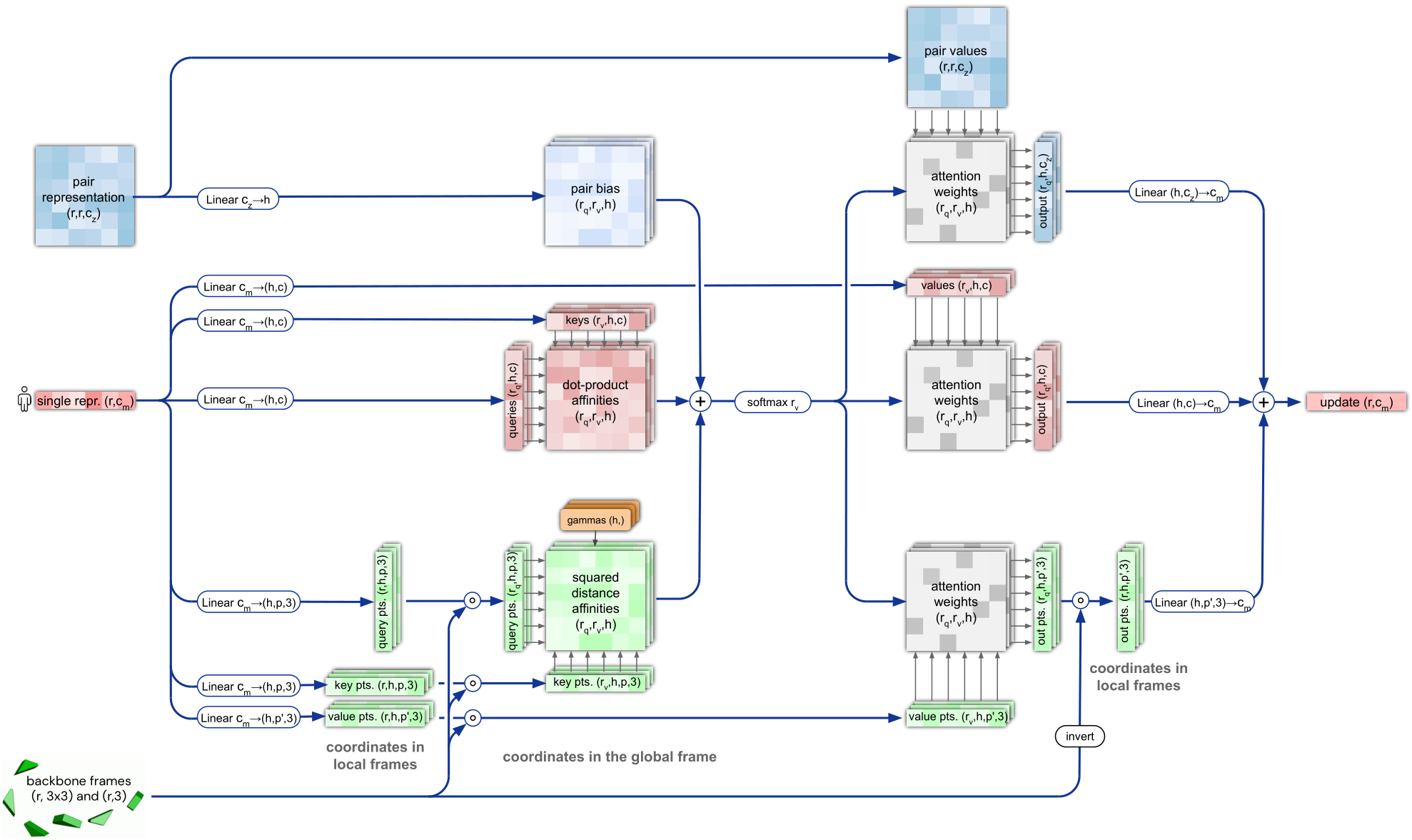
其主要由三个部分组成,蓝色的表示Evoformer输出的pair-repr.,红色的表示目标序列的表示,绿色的表示结构的内坐标表示。其图上展示的很清楚,三个部分共同计算attention,并用于更新目标序列的表示。
比较特别的是其Invariant的attention计算,如下:
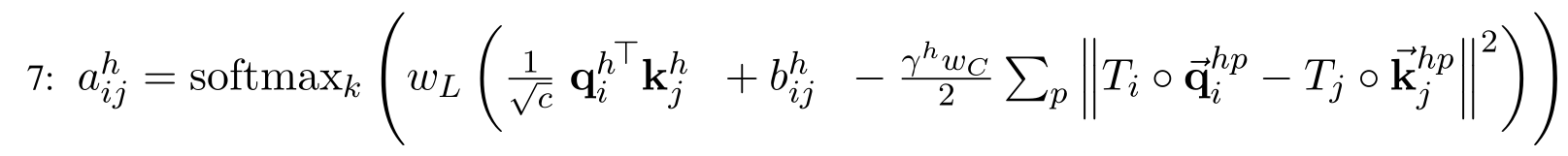
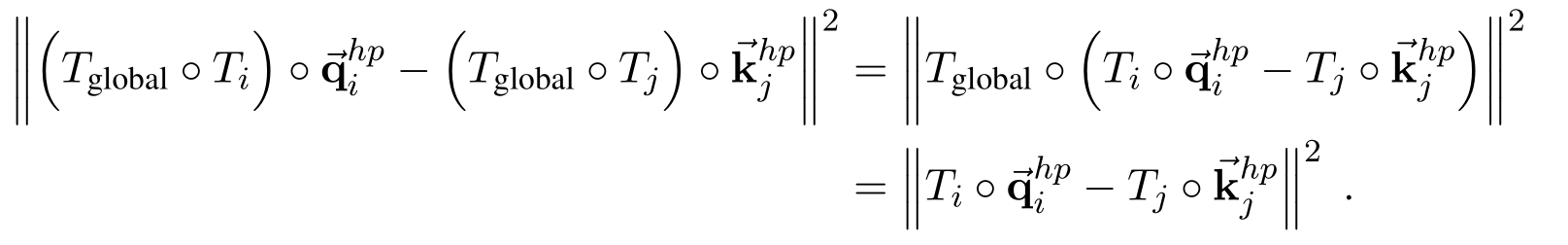
这里需要先提下最终可视化蛋白结构的方法,其每个残基用一个$ T $表示, $ T = (R, \widetilde{t}) $, $ R $ 是一个$ [3,3] $的旋转矩阵, $ \widetilde{t} $ 是一个 $ [3] $ 的平移矩阵, 所以蛋白每个残基的结构可以化作为 $ T $ 的表示, 在初始化时, 其全部 $ T $ 为 $ 0 $ , 每次都根据现有的pair-repr., 目标序列的表示及当前 $ T $ 来更新预测需要更新的 $ T $ , 将预测的 $ T $ 乘于当前 $ T $ 之上。而这个attention的计算方式保证了其在旋转或平移蛋白后, 不会对attention的计算结果产生变化,即3D等变性。
更新后的目标序列的表示使用linear来直接预测backbone和sidechain的各个内坐标的 $ T $ 更新。
Loss
其loss如下:
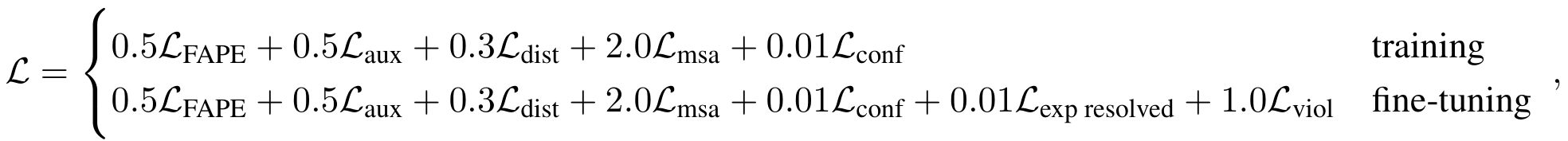
| Loss组成 | 内容 |
|---|---|
| FAPE | Frame aligned point error。预测与真实结构aligned后,所有原子预测位置与对应位置距离差。 |
| aux | auxiliary loss。这是Cα的FAPE及内坐标的loss,是在推断模块每次更新都计算后取平均。 |
| dist | 两两残基分箱距离预测(接触距离预测)。 |
| msa | masked MSA的预测。 |
| conf | model confidence loss(预测残基置信度)。 |
| exp resolved | 是否被高分辨率解析。 |
| viol | Structural violations。根据简化力场加入的约束,防止有过于离谱的原子间距离或角度。 |
训练方式
训练采用MC(蒙特卡洛)的方式,每次选一个cycle进行梯度回传。recycle和这样MC的训练方式都是减少计算开销的方式。这样还有个好处就是可以均匀的训练到每个cycle的情况。
小结
AF2采用了少量的原创内容,但每一步都感受到了精细的打磨,这也是为什么AF2有相当多的作者,每个作者都精细维护其负责的部分。汇聚如此多的人才使用了包含领域内几乎所有的数据,同时,使用了大量TPU,如此的资源几乎不是单独课题组可以匹敌的(哭)。所以这也不难理解为什么他能比之前的模型效果超出这么多了。不过如此深度的训练出来的模型,其应该一定估计会比基于物理化学的方法有更多的bias或过拟合,等到十年后更多的数据出来,或许可以发现AF2的许多不足之处。
本文由 KoN 创作,如果您觉得本文不错,请随意赞赏
采用 知识共享署名4.0 国际许可协议进行许可
本站文章除注明转载/出处外,均为本站原创或翻译,转载前请务必署名
原文链接:/archives/alphafold2阅读笔记
最后更新:2023-11-27 02:29:32


